



1.单基因遗传病
(1)常染色体显性遗传疾病
常染色体显性遗传病是位于常染色体上的显性致病基因引起的,由于它的特别性,所以常常具有以下特点:
①只要体内有一个致病基因存在,就会发病。双亲当中有一个是患者,就会遗传给他们的子女,子女中半数可能发病。若双亲都是患者,其子女有2/3可能发病(患者均为杂合体),若患者为致病基因的纯合体,子女便会全部发病。

肤色——显性遗传
②此病与性别无关,男女发病的机会均等。
③在一个患者的家族中,可以连续几代出现此病患者。但有时因内外环境的改变,致病基因的作用不一定表现(外显不全),一些本应发病的患者可以成为表型正常的致病基因携带者,而他们的子女仍有1/2的可能发病,出现隔代遗传。
④无病的子女与正常人结婚,其后代一般不再有此病。常染色体的遗传病主要有以下几种类型:
家族性高脂蛋白血症
血浆中一种或多种脂蛋白异常的现象所引起的疾病即为高脂蛋白血症。高脂蛋白血症有原发性和继发性两类,原发性患者多为遗传的。高脂蛋白血症在生物化学上可分5型,分别为:
血浆
Ⅰ型,高乳糜微粒血症;
Ⅱ型,高β脂蛋白血症;
Ⅲ型,宽β型高脂蛋白血症;
Ⅳ型,高前β脂蛋白血症;
Ⅴ型,高前β脂蛋白及乳糜微粒血症。
家族性高胆固醇血症
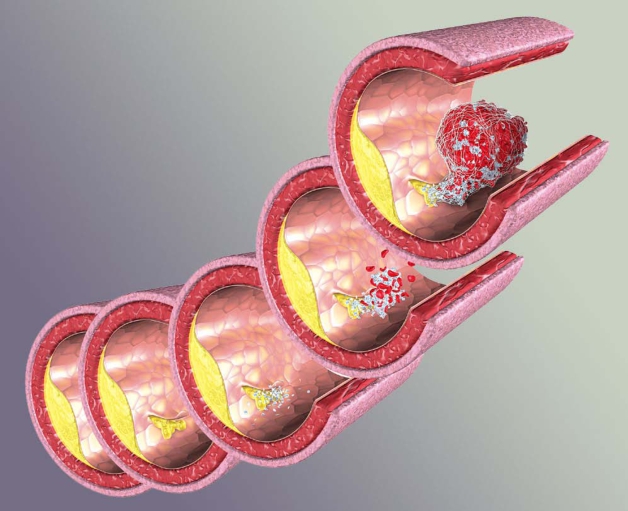
动脉粥样硬化性血栓形成过程
动脉粥样硬化脂纹脂斑
其中Ⅱ型及Ⅲ型与冠状动脉粥样硬化性心脏病关系最密切。高脂蛋白血症Ⅱ型患者的生化特点是血浆中β脂蛋白大量增加,胆固醇和磷脂也增加,甘油三脂正常或微增。这些胆固醇和脂质在动脉管内沉着,内膜呈现局限性增厚,形成斑块,然后发生崩溃,形成溃疡和软化,分解出一种黄色粥样物质,故称“粥样硬化”。此后有纤维组织增生,并可有钙质沉着,或发生局部血栓形成,内膜凹凸不平,管腔狭窄,使管壁硬化。若硬化发生于大动脉,对血液供应不会产生影响;若发生于中型动脉(如冠状动脉、脑动脉、肾动脉等),则引起相应脏器供血不足,甚至发生梗阻。所以高脂蛋白血症Ⅱ型的最危险并发症是早发的冠状动脉粥样硬化,常并发心绞痛与心肌梗死。
马尔芬氏综合征
此病也叫蜘蛛指症。致病基因携带者可在儿童少年期发病,也可在青春期或成年的早、晚期发病。患者一般身材较高,四肢细长,脊柱后侧凸,关节松弛,胸部凹陷或突起,两臂伸开长度大于身高,脚、手大,指(趾)细长,头长;肌肉系统发育较差,皮脂少;眼部有晶体上颞部半脱位,虹膜震颤,近视,自发性视网膜剥离;患者60%~80%有心血管系统疾病,如二尖瓣机能障碍、主动脉瘤、肺动脉中层变性伴发破裂,房室间隔缺损等。美国著名女排选手海曼,身高1.96米,四肢修长,近视。在比赛中因血管瘤破裂而死,最后专家确诊为马尔芬氏综合征。
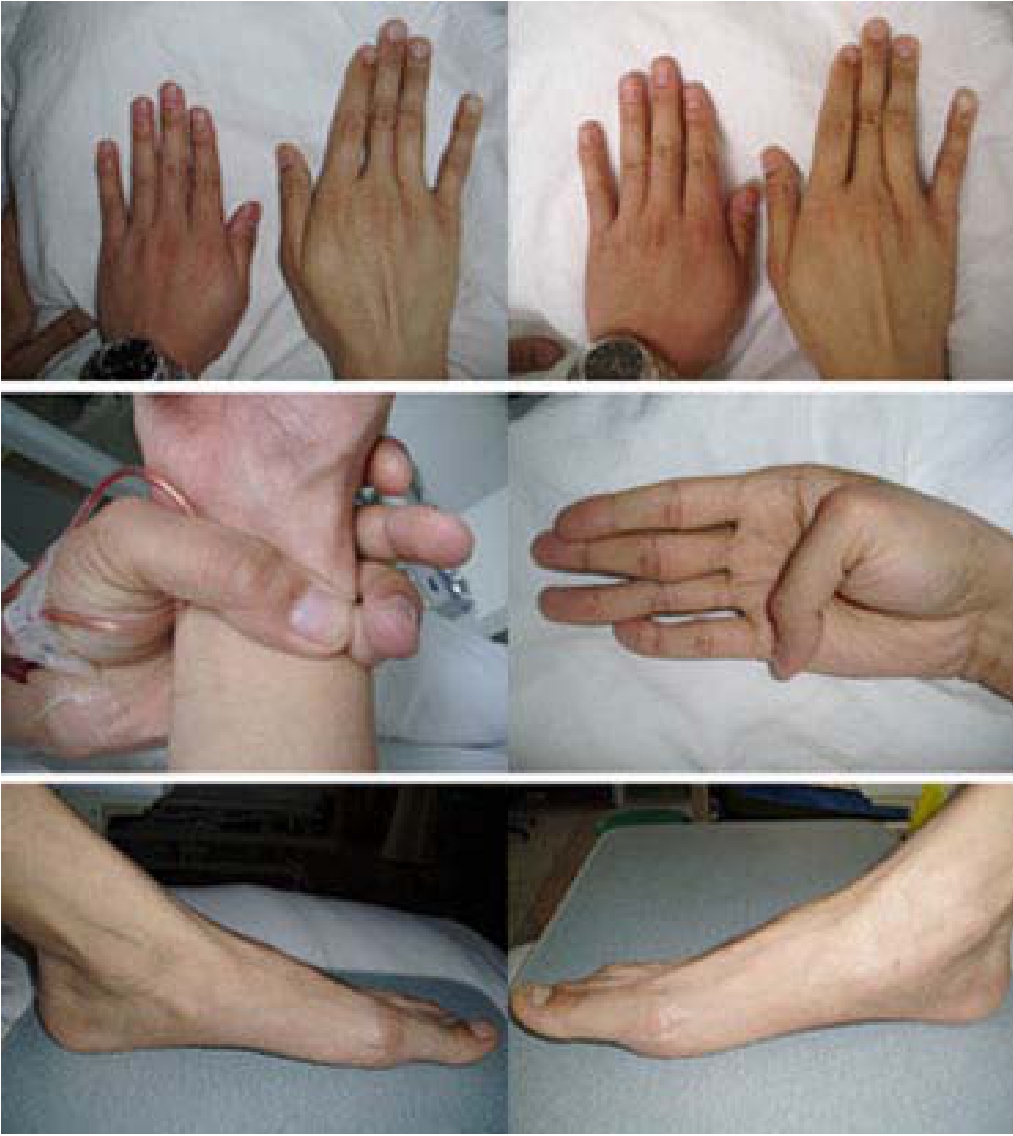
蜘蛛指症
威尔逊氏综合征

从左至右依次显示为正常肝脏、脂肪肝、肝硬化
由于基因突变引起患者体内铜代谢障碍所致。致病基因携带者在10岁之前发育一切正常,往往在10~20岁之间突然发作,会出现脑中心退化,肝细胞被纤维组织代替从而造成肝硬化,角膜中间出现色环,眼球震颤,肌张力亢进,尿液中含有大量末端双羧氨基酸的肽和氨基酸残基等。
亨丁顿氏舞蹈病
此病是一种完全符合孟德尔氏遗传的显性遗传病。患者20岁前很少发病,20岁后发病率逐渐增高。发病时,最初表现为情绪波动,随后出现舞蹈性动作,癫痫发作,体力和智力不断减退,进行性痴呆。常于症状出现后的4~20年间死亡。此病有明显的家族遗传史,只要双亲之中有一个是患者,他们的子女中的发病概率至少有1/2。
亨丁顿氏舞蹈症病毒
结肠息肉
此病有明显家族遗传倾向,患者最早可在20岁左右发生恶变,结肠上长有大小不等的肉瘤,引起胃肠出血和腹泻,息肉恶变的可能性较大,需进行结肠切除手术。同此病相类似的另一种肠道遗传病是空肠息肉,患者的早期在口、唇周围及口腔黏膜和手指上会出现色素斑点,到成年就会出现消退的趋势。良性息肉主要分布在空肠,但也偶发在肠道的其他部位,或膀胱及呼吸道,伴有腹部绞痛、胃肠出血和肠套叠等并发症。在儿童期可发病。
结肠息肉手术
阵发性心动过速
此病有明显的家族史,可连续几代遗传。可在任何年龄阶段出现阵发性心动过速,病理多属器质性,也有纯功能性的。
定时进行血压测量
体质性低血压
此病患者往往是一些体质瘦弱的人,女子要比男子多。多数患者无自觉症状,少数有疲倦、健忘、头晕、头痛等。这些症状常因某些疾病或营养不良所致。
椭圆形红细胞增多症

遗传性椭圆形红细胞
此病是由两个不同位点上的基因各自控制的疾病,患者有50%或更多的红细胞呈椭圆形、卵圆形、香肠形和杆状(正常人最多为10%),这种异形红细胞最早可出现在3~4个月龄婴儿的外周血液循环中。此病患者在儿童少年多无症状表现,但存在不同程度的溶血,而且病人有肝、脾肿大、腹水、黄疸等症状。
肌强直性营养不良
此病的遗传表现为母亲如果有此病,其子女患病的可能性也就越大。有的患者在儿童期发病,而更多的是在成年初期发病。最常见的症状是颌部和手部肌肉松弛、收缩困难,肌肉萎缩、无力,面容无表情,而且还可发生白内障。男性有额秃,睾丸萎缩;女性则有闭经,痛经和卵巢囊肿。尚可有胃肠道平滑肌功能障碍,部分病人智力衰退甚至痴呆。
肌肉萎缩的手
先天性肌强直
肌强直患者
此病主要症状为普遍性肌强直和肌肥大,多数在出生时或儿童早期即发病,少数至青春期发病。患者肢体僵硬,动作笨拙,静止不动后或在寒冷环境中症状加重,反复运动可暂时减轻症状。坐或站立一段时间后,不能立即起立或起步。突然受惊吓时,可引起全身肌肉的强直性收缩;跌倒时不能将手伸出撑住地面及时爬起;与人握手后要较长时间才能松开。打喷嚏后,双眼仍紧闭;发笑后,面部表情肌不能及时恢复;温暖的环境能使肌强直减轻。症状严重程度可因人而异,最轻者甚至无自觉主诉,仅在家系调查中发现。个别病人在肌肉多次收缩后症状不见减轻,反而加重,称为反常性肌强直。病人全身肌肉发育良好,常伴肥大。此病预后良好,多数随年龄增长而症状减轻,对寿命并无影响。
周期性麻痹
此病根据致病基因携带者受刺激后机体的血钾水平的不同表现可以分为以下不同种类。一种类型是在激烈运动后长时间休息、食用高碳水化合物、焦急忧虑、遇到寒冷或服用多种药物(包括胰岛素、肾上腺素、乙醇、一些无机物、皮质激素和甘草属植物)等情况下,可促发机体低血钾麻痹。而另一种类型是激烈运动时、服用氯化钾以及使用某些麻醉剂可引起高血钾性麻痹。
周期性瘫痪疾病病因
胱氨酸尿症
此病主要是由肾小管对胱氨酸、赖氨酸、精氨酸、鸟氨酸的重吸收发生障碍所致。患者尿中有上述4种氨基酸排出,但无任何症状。由于胱氨酸易生成六角形结晶,故可发生尿路结石(胱氨酸结石)。尿路结石可引起尿路感染和绞痛。纯合子患者4种氨基酸排泄量均可增加,杂合子患者胱氨酸和赖氨酸排泄量仅有少量增加。
遗传性球形细胞增多症
此病是一种慢性溶血性贫血,主要特征是黄疸、脾大、红细胞球形改变、脆性增加。新生儿期发病时可有严重贫血和黄疸症状,婴儿期除轻度或中度贫血外往往不会出现其他症状,幼儿及年长儿发病时,其主要症状是轻度黄疸及贫血。如过度劳累、受冷感冒可使黄疸加重,而且还伴有发热、呕吐、腹痛、肝脾压痛、无力、心跳加快、气促,脾脏增大可达肋下 2.8厘米,肝脏略有增大。在血液学检查中,可以看见红细胞直径变小,胞体变圆,网织红细胞增高5%~20%,红细胞脆性增高可达0.40%~0.68%,自身溶血试验呈阳性。
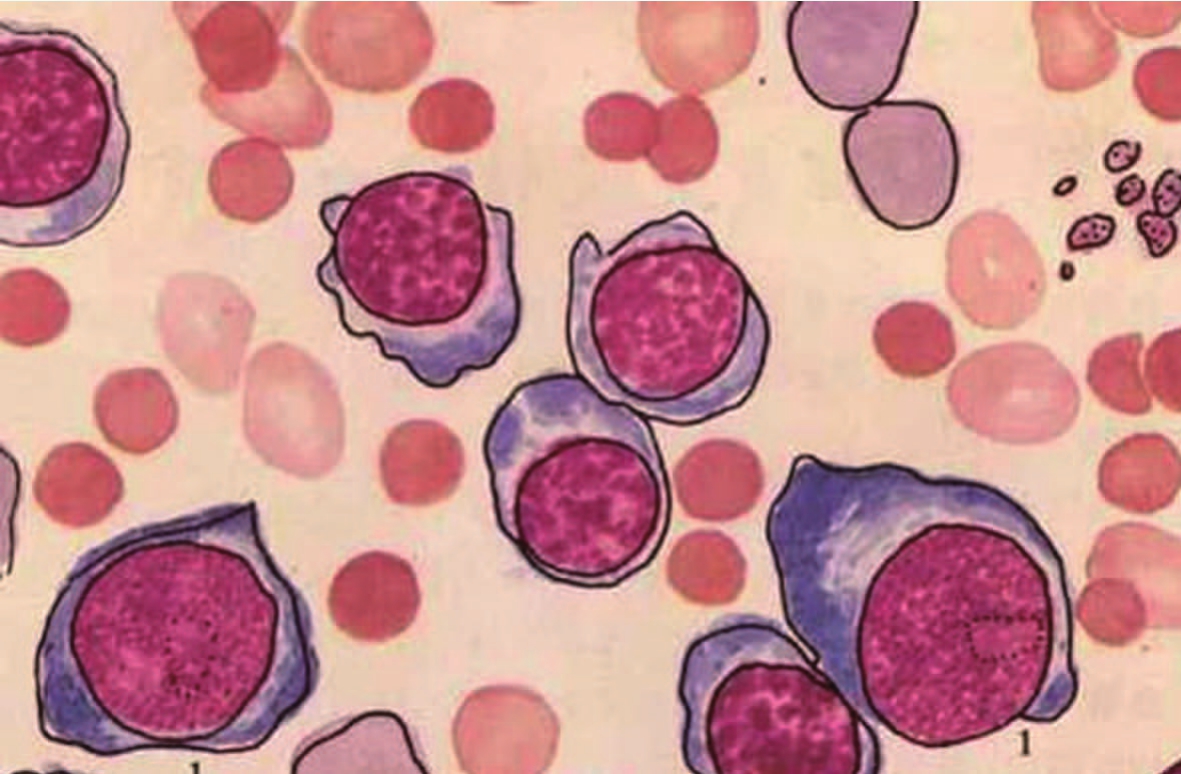
遗传性球形红细胞增多症骨髓象
常染色体显性遗传病的种类很多,除此之外,比较常见的还有软骨发育不全症、短指畸形、肾性糖尿病、先天性白内障、夜盲症、青光眼、视网膜母细胞瘤、先天性眼睑下垂、多指畸形、多囊肾、遗传性神经性耳聋、过敏性鼻炎、牙齿肥大症、多胎妊娠及尿崩症等。
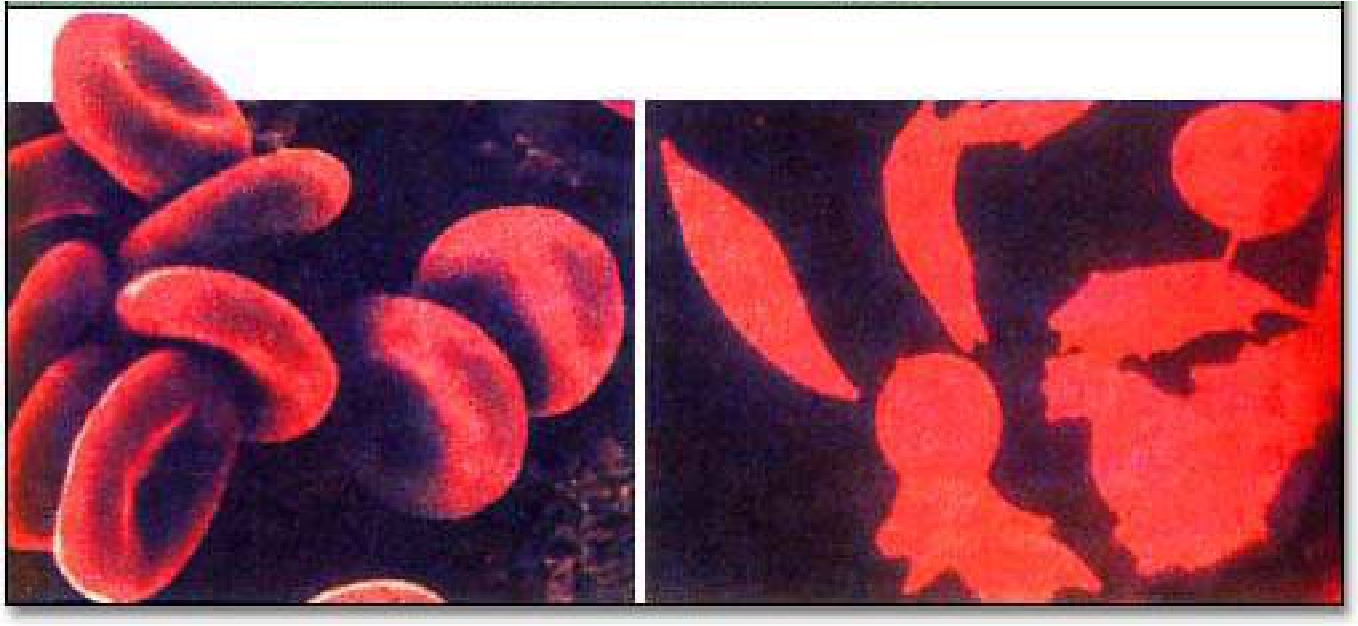
正常型红细胞(左)与镰刀形细胞贫血症红细胞(右)的形状比较
免责声明:以上内容源自网络,版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。










